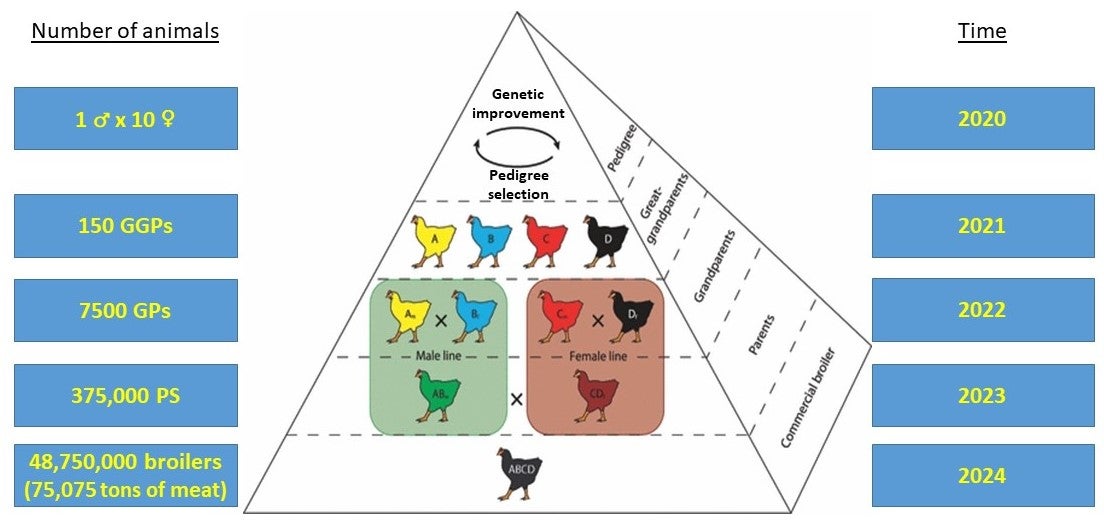
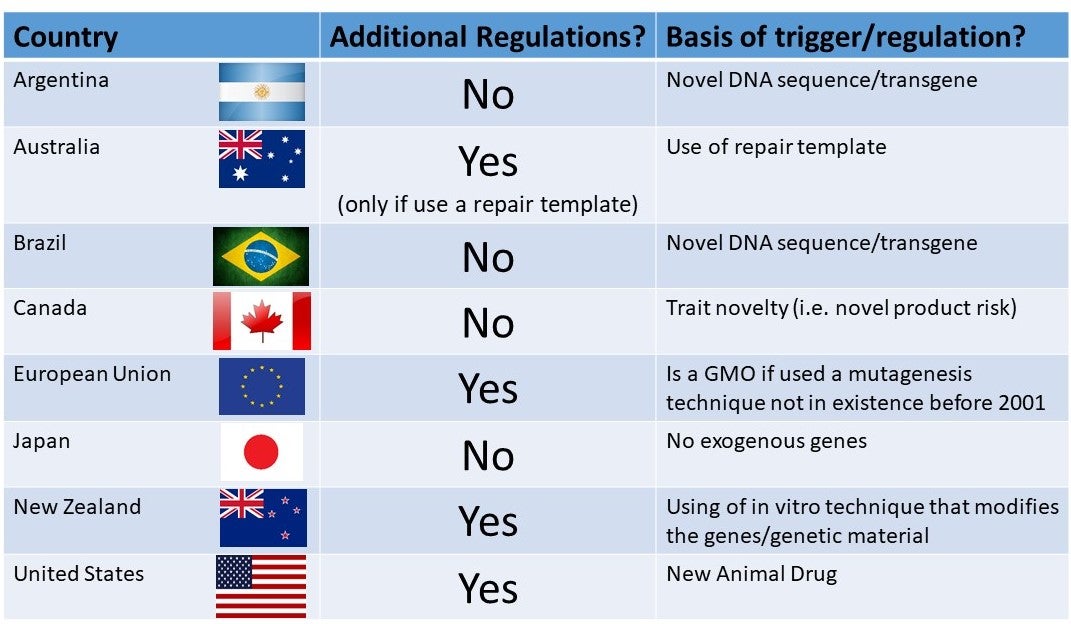
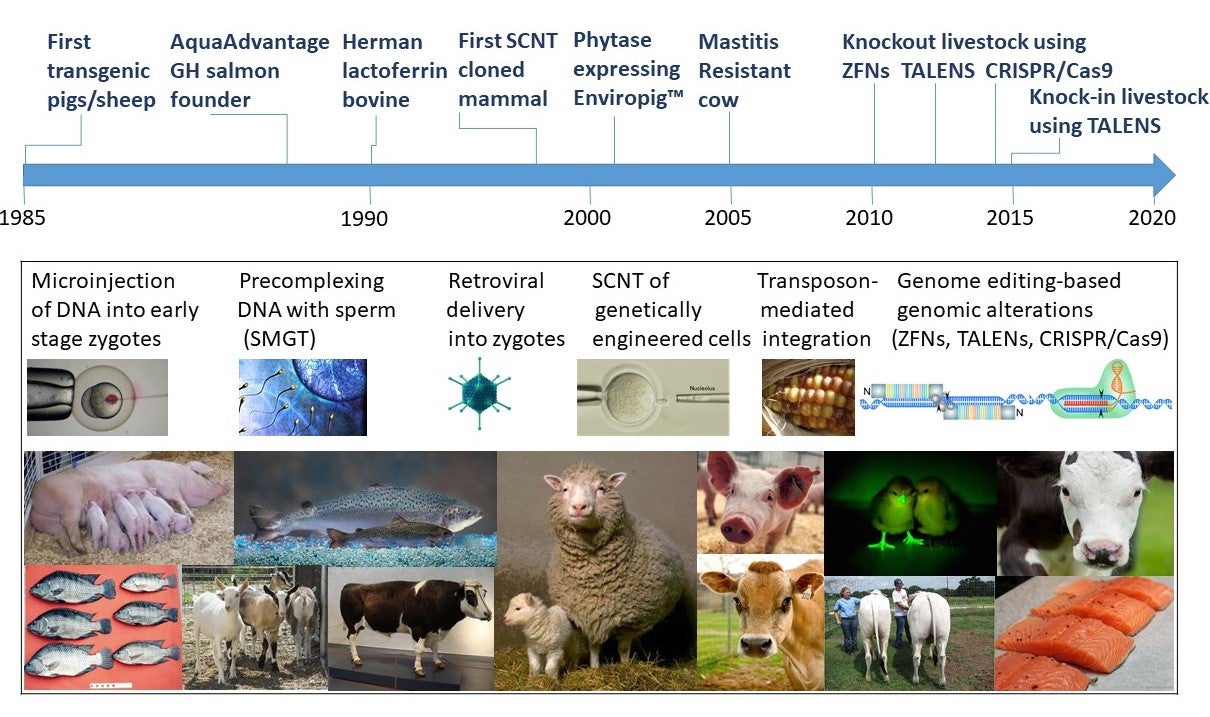
This is part 2 of a 1, 2, 3 part series on Genetic Engineering in Livestock
When AquaBounty sought to commercialize the first transgenic food animal in the mid 1990s (first produced in 1989), there was no official regulatory approach in place. Former CEO of AquaBounty technologies Dr. Ron Stotish in a 2012 abstract entitled “AquAdvantage salmon: pioneer or pyrrhic victory” (Transgenic Research 21: 913-914) wrote :
“AquaBounty consulted FDA and other government agencies in hopes of identifying a regulatory process that could be employed to review and approve the AquAdvantage salmon for food use in the United States. AquaBounty established an Investigational New Animal Drug [INAD] file with the Center for Veterinary Medicine in 1995, well in advance of any clear regulatory paradigm. Between 1995 and 2009, the sponsor [AquaBounty] conducted a variety of GLP studies aimed at meeting what was hoped to be the eventual regulatory requirement for an application of this nature. Although there was informal consultation and communication between the sponsor and CVM staff during this time, it was not until 2009 that CVM [FDA Center for Veterinary Medicine] released Guidance Document 187, codifying requirements for consideration of an application for a transgenic animal.”
This 2009 Guidance Document 187 was entitled “Regulation of Genetically Engineered Animals Containing Heritable rDNA Constructs”. The Federal Food Drug and Cosmetic Act (FFDCA), defines a drug as an “article intended for use in the diagnosis, cure, mitigation, treatment, or prevention of disease in man or other animals;” and “articles (other than food) intended to affect the structure or any function of the body of man or other animals.” A “New Animal Drug includes a drug intended for use in animals that is not generally recognized as safe and effective for use under the conditions prescribed, recommended, or suggested in the drug’s labeling, and that has not been used to a material extent or for a material time.” Using this definition the FDA considered the “regulated” article to be “the rDNA construct in a GE animal that is intended to affect the structure or function of the body of the GE animal, regardless of the intended use of products that may be produced by the GE animal.”
In that guidance the FDA defined GE animals as animals with both heritable and non-heritable rDNA constructs. The classification of transgenes in animal genomes as drugs meant that each animal lineage derived from a separate transformation event (or series of transformation events) was considered to be a separate new animal drug, subject to a separate new animal drug approval. This determination meant that all unapproved GE animals, their offspring, and their food products were “deemed unsafe”. The FDA exercised enforcement discretion for GE animals of non-food-species that are raised and used in contained and controlled conditions such as GE laboratory animals used in research institutions. In other words, researchers working with the literally millions of GE laboratory animals did not have to go through the INAD and NADA (New Animal Drug Application) requirements.
That left the small group of mostly public sector agricultural researchers working with food animals saddled with the requirements needed to get a new veterinary drug approved if any of their work was to ever reach the market. These requirements include a seven-step regulatory process in which the agency examines the safety of the recombinant DNA (rDNA) construct to the animal, the safety of food from the animal, and any environmental impacts posed (collectively the “safety” issues), as well as the extent to which the performance claims made for the animal are met (“efficacy”).
Molecular characterization of the rDNA construct determines whether it contains DNA sequences from viruses or other organisms that could pose health risks to the GE animal or to those eating the animal. Molecular characterization of the GE animal lineage determines whether the rDNA construct is stably inherited over multiple generations. Phenotypic characterization assesses whether the GE animals are healthy, whether they reach developmental milestones as non-GE animals do, and whether they exhibit abnormalities. A durability assessment reviews the sponsor’s plan to ensure that future GE animals of this line will be equivalent to those examined in the pre-approval review. If the GE animal is intended as a source of food, FDA assesses whether the composition of edible tissues differs and whether its products pose more allergenicity risk than non-GE counterparts.
In the meantime, all investigational GE animals, their littermates, and surrogate dams and their products were deemed “unsafe” and had to be disposed of by “incineration, burial, or composting.” My colleague, Dr. Matt Wheeler, at the University of Illinois has been working on transgenic pigs for over twenty years. His work has included expressing the bovine lactalbumin gene in the milk of transgenic pigs (1998), which enhances lactation performance and preweaning piglet growth rates (2002). This work has been carried out under an INAD, which initially authorized the rendering of a certain subset of experimental animals. A misunderstanding over the regulatory status of cogestating one INAD line of transgenic pigs with littermates of another INAD line of transgenic pigs resulted in the FDA determining that Dr. Wheeler had “failed to follow protocol”. One Friday, Dr. Wheeler relayed to me, that the FDA swept down on his laboratory and confiscated his computer and laboratory note books. They also locked him out of his own laboratory for several days,
The experience he says, “made him do some soul searching as to whether he wanted to continue doing work with transgenic livestock.”
Such actions have done little to build trust with the regulated academic community. Dr. Wheeler estimates he has incinerated thousands of transgenic pigs and their littermates and surrogate dams as unapproved new animal drugs during the course of his research, and guesses the added costs of being under INAD requirements to be in the vicinity of USD$2 million. Fortunately academic research institutions and small companies are exempt from additionally paying the recurring annual INAD review fee, which can be thousands of dollars.

The genetically modified AquAdvantage salmon (top) gets to market weight in half the time required for its traditional counterpart. AquaBounty.
To continue on with the AquAdvantage saga, Dr. Stotish wrote in 2012 “by mid 2010 [AquaBounty] had concluded all the necessary research, submitted all required regulatory studies, and received the results of the CVM reviews indicating satisfaction with the submitted data in addressing all requirements for approval. The Center convened a Veterinary Medicine Advisory Committee [VMAC] to review the results of the CVM assessment and conclusions on September 20, 2010. ” Little did the company know that there would be a 5 year delay and to complete “radio silence” from the FDA following the 2010 VMAC meeting until approval in November 2015. The salmon has had a long road to market.
The VMAC meeting for the AquAdvantage salmon was held in September 2010. During the course of the meeting the VMAC members discussed the strengths and weaknesses of the data (4). Ultimately the consensus document of the VMAC charged with providing advice and recommendations to the FDA found (1) “no evidence in the data to conclude that the introduction of the construct was unsafe to the animal,” (2) that the studies selected to evaluate whether or not there was a reasonable certainty of no harm from consumption of foods derived from AquAdvantage salmon were “overall appropriate and a large number of test results established similarities and equivalence between AquAdvantage Salmon and Atlantic salmon,” and (3) that the AA salmon did grow faster than their conventional counterparts.
The potential environmental impacts from AA salmon production were mitigated by the proposed conditions of use which limits production to FDA-approved, physically-contained fresh water culture facilities. The eyed-egg production site is located on Prince Edward Island (Canada), and the grow-out of market fish is proposed to occur in Panama with multiple biological (all female, triploid), physical (land-based tanks with fencing/screening), and geographical (hydroelectric dams with no fish passage, thermal-lethal downstream temperatures) redundant containment measures. The VMAC concluded that although they “recognized that the risk of escape from either facility could never be zero, the multiple barriers to escape at both the PEI and Panama facilities were extensive”.
Activist disinformation campaign

Disinformation that was created to misinform about the nutritional data of the AquAdvantage salmon
The data package that AquaBounty voluntarily made publicly available in advance of the 2010 VMAC in a good-faith attempt to increase transparency in the regulatory process, was used by special interest groups like the Center for Food Safety who misrepresented the data by suggesting that “FDA’s 2010 data release showed that GE salmon have 40% higher levels of the growth hormone IGF-1, which increases the risk of cancer. And finally, wild salmon has 189% higher levels of omega-3 fatty acids than GE salmon can produce.” This false interpretation stands in stark contrast to the FDA finding which was “AquaBounty (ABT) salmon meets the standard of identity for Atlantic salmon as established by FDA’s Reference Fish Encyclopedia. All other assessments of composition have determined that there are no material differences in food from ABT salmon and other Atlantic salmon.” This type of vilification of technology and fearmongering by special interest groups with no interest in truthfulness does nothing to benefit society.
The U.S. Food & Drug Administration ultimately approved AquAdvantage salmon for sale in November 2015. But an obscure rider was attached to a budget bill by Alaska senator Lisa Murkowski in December of that same year, effectively blocking the FDA from allowing salmon into the U.S. In the meantime Canadian regulatory authorities approved the fish in 2016, and sold 5 tons of fillets (4.5 metric tons) there in 2017, and the same in 2018. “The people who bought our fish were very happy with it,” Stotish is quoted in a press article. “They put it in their high-end sashimi lines, not their frozen prepared foods.”
It is worth noting that the US imported 339,000 metric tons of salmon in 2016, worth more than $3 billion. The vast majority of that came from farmed Atlantic salmon raised in floating cages off the coasts of Canada, Chile, Norway and Scotland, and flown to the United States. Atlantic salmon raised in land-based tanks like the AquAdvantage salmon is rated “best choice” by Seafood Watch, and the carbon footprint of salmon produced in land-based closed systems is <50% lower than that of salmon produced in conventional marine net pen fish farms in Norway and delivered to the U.S. by air.
In 2018, FDA approved an application by AquaBounty for a growout facility in Indiana, offering an opportunity to produce US-raised Atlantic salmon. And finally on March 8, 2019, 30 years after the founder fish was produced in Canada in 1989, the FDA deactivated this import alert pertaining to the GE salmon. It still has not reached US consumers, despite recent court victories.
AquaBounty estimated it has spent $8.8 million on regulatory activities to date including $6.0 million in regulatory approval costs through approval in 2015, $1.6 million (and continuing) in legal fees in defense of the regulatory approval, $0.5 million in legal fees in defense of congressional actions, $0.7 million in regulatory compliance costs (~$200,000/year for on-going monitoring and reporting including the testing of every batch of eggs), not to mention the $20 million spent for maintaining the fish while the regulatory process was on-going from 1995 through 2015 (David Frank, AquaBounty; pers. comm; January 2020).
Meanwhile
While the AquAdvantage fish has been awaiting regulatory approval, salmon breeders in Norway have been busy selecting for fast-growing salmon. The genetic gain for growth-rate in salmon has been estimated at 10–15% per generation. Farmed salmon have been exposed to ≥12 generations of domestication and was the first fish species to be subject to a systematic family‐based selective breeding program which began in 1975. Studies on farmed salmon in the 9-10th generation of selection showed their size relative to wild fish was 2.9:1 under standard hatchery conditions, and 3.5:1 under hatchery conditions where growth was restricted through chronic stress. In other words, selective breeding programs have produced genetically-distinct lines of fast-growing salmon, and since the 1970s, tens of millions of these fertile farmed salmon have escaped into the wild. Glover estimated that over three to four decades, introgression of farmed salmon into Norwegian wild salmon populations ranged from 0% to 47% per population, with a median of 9.1% .
Presumably these fast-growing salmon pose the same risks as the AquAdvantage salmon, although conventional breeding is not regulated and the latter was associated with decades of delay, millions of dollars in new animal drug regulatory approval costs, and all fish must be raised as sterile triploid females in land-based containment tanks and is subject to ongoing regulatory compliance costs.
This might sound like an argument to just let conventional breeding do its job. As a geneticist I have to agree, and this is absolutely an option, albeit a slower one, for traits that vary in the target population. But for some traits like disease resistance, having the ability to bring in useful genetic variation enables breeders to introduce novel traits that cannot be achieved by conventional breeding. The problem is that it has not be possible to bring transgenic food animals to market, even those that address traits of interest to the consumer like disease-resistance and animal welfare traits. All of it is blocked.
Having a different regulatory standard triggered by the process (i.e. rDNA versus other breeding methods) used to make a product , rather than on the risk associated with the product itself runs counter to the The United States “Coordinated Framework for the Regulation of Biotechnology,” which is technically agnostic towards the technology or process under review. According to the Office of Science and Technology Policy (OSTP) in the Federal Register in 1992:
“Exercise of oversight in the scope of discretion afforded by statute should be based on the risk posed by the introduction and should not turn on the fact that an organism has been modified by a particular process or technique … (O)versight will be exercised only where the risk posed by the introduction is unreasonable, that is, when the value of the reduction in risk obtained by additional oversight is greater than the cost thereby imposed ”
This suggests that the U.S. should only exercise regulatory authority over organisms — plant or animal — based on the risks they pose. This is irrespective of the breeding technique used to produce them, and used only when the risk posed is unreasonable, which is clarified to mean the cost of regulatory oversight is not greater than the reduction in risk obtained by that oversight. it is difficult to see how this applies to transgenic animals given the history of the fast-growing salmon. It was hoped that new breeding methods like gene-editing would be treated more like conventional breeding. Unfortunately, that is not how it happened, as discussed in Part 3 of this series.