The email was simple enough. It was a request from a member of the press asking “I would appreciate your reaction/comments to the recently published study on GMO corn for an article I am putting together on it. Deadline: Wednesday 4 January.”
Just when I thought I was going to get a day off to myself to write up my own research results, in comes the dreaded time-sensitive press request for comments on a recently published paper. Dreaded because to respond properly means I need to sit down and read the whole paper and ensure I have understood the materials and methods, results, and discussion. For me that is a commitment of a couple of hours. And to top things off – it was a paper by Mesnage from France’s infamous Séralini group whose previous works have had numerous flaws. But I made a New Year’s Resolution to be more active in critiquing agricultural science and can’t in good faith renege on that resolution on January 2nd.
The paper’s title “An integrated multi-omics analysis of the NK603 Roundup-tolerant GM maize reveals metabolism disturbances caused by the transformation process” suggested the researchers had uncovered some altered metabolic processes caused by the transformation process used to create the NK603 Roundup-tolerant genetically modified (GM) maize line. This event was achieved by direct DNA transformation by microparticle bombardment of plant cells with DNA-coated gold particles and regeneration of plants by tissue culture on selective medium. This transformation process presumably happened last century as the feed/food approval for this line in the United States occurred in 2000. However, upon reading the abstract the paper was nothing to do with disturbances caused by the transformation process, but rather it was about whether the product of this transformation event was “substantially equivalent” based on proteomics and metabolomics evaluation. Strangely, the “conclusiony”-sounding title of the paper therefore had nothing to do with the experimental design or findings discussed in the paper.
According to the results section, the actual “objective of this investigation was to obtain a deeper understanding of the biology of the NK603 GM maize by molecular profiling (proteomics and metabolomics) in order to obtain insights into its substantial equivalence classification.” In plain English – the intent of the paper was to examine both proteins and metabolites found in NK603 Roundup-tolerant GM maize (both treated and untreated with Roundup), and non-GM isogenic lines to determine if the three groups were substantially equivalent using sensitive “-omics” assays.
To perform such an evaluation requires a common agreement as to what substantial equivalence means, and what constitutes an appropriate comparator(s). Unfortunately, not such common understanding exists. According to an OECD publication in 1993, substantial equivalence is a concept which stresses than an assessment of a novel food, in particular one that is genetically modified, should demonstrate that the food is as safe as its traditional counterpart. This has been interpreted to mean that the levels and variation for characteristics in the genetically modified organism must be within the natural range of variation for those characteristics considered in the comparator.
And this brings up the issue of an appropriate comparator. Typically this involves the comparison of key compositional data collected from both the recombinant-DNA crop plant and the isogenic non-GM counterpart, grown under near identical conditions. Ideally, conventional non-GM corn hybrids are also included in analyses to determine the level of natural variation for compositional data in conventional varieties that are considered to be safe for consumption based on a history of safe use.
According to the original studies of the NK603 GM maize variety compositional analyses were conducted on the key corn tissues, grain and forage, produced in multiple locations (Kansas, Iowa, Illinois, Indiana, and Ohio in 1998 and in trials in Italy and France in 1999). Grain and forage samples were taken from plants of the corn event NK603 and the non-modified control both years. In the E.U. field trials, reference grain and forage samples also included 19 conventional, commercial hybrids. The NK603 plants were treated with Roundup Ultra herbicide. Fifty-one different compositional components were evaluated.
Not surprisingly there are protocols on how to best carry out experiments on GM crops that are accepted by regulatory agencies world-wide (OECD 2006; Codex 2009). According to EFSA, for compositional analysis risk assessment, field trials will include: the GM plant under assessment, its conventional counterpart (isogenic non-GM counterpart), and non-GM reference-varieties, representative of those that would be normally grown in the areas where the field trials are performed. The later puts some figures and context to the natural biological variation in the different plant varieties we commonly consume.
So what did the Mesnage paper in question do? The researchers planted a single replicate of the GM plant under assessment (DKC 2678 Roundup-tolerant NK603) and its conventional counterpart (DKC 2575 – although the exact genetic makeup of this line and whether it is a true isogenic counterpart is not well elaborated in the paper) at a single location on two different years. Half of the GM plants each year were treated with Roundup. Then the corn kernels were harvested and the proteins and metabolites from the three groups were assayed using proteome and metabolome profiling, the data from the two years were merged and analyzed. The three groups (isogenic non-GM counterpart), GM plant without roundup treatment, and GM plant with roundup treatment separated into three distinct groups based on a principal component analysis (PCA).
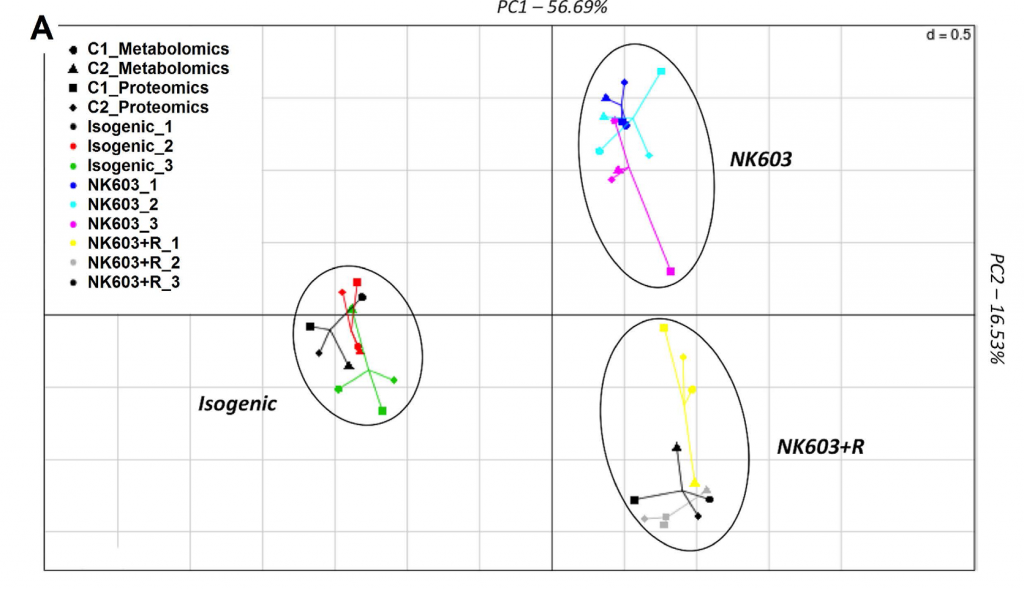
Integration of metabolome and proteome profiles of the NK603 maize and its near-isogenic counterpart into a multiple co-inertia analysis projection plot.
I draw your attention to a very similar graph (below) in a paper I recently published which shows a PCA analysis of the transcriptome (genes expressed) from cattle that have been exposed to different viruses and bacteria. Basically PCA can pull apart patterns of gene expression in different groups of cattle in response to the specific environmental challenges they are facing. The controls can clearly be seen to be clustering down in the bottom right corner, and the bacterial infections tend to cluster to the right and differently than those infected with viruses which cluster to the left.
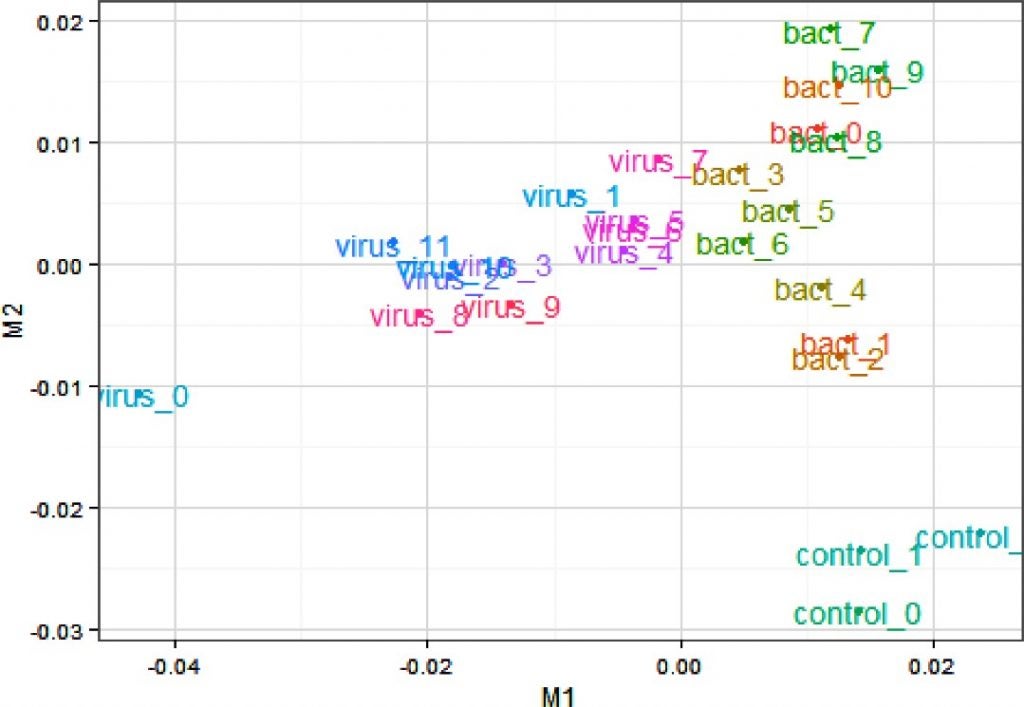
Multidimensional scaling plot of samples based on all genes
That is – if you expose plants or animals to different environmental or disease challenge conditions – they express different genes in response. That is typically why researchers do “–omics” studies, to try to identify which genes/proteins/metabolites respond to different environmental conditions. What they do not show is whether any of beef that might be derived from these animals would be unsafe to eat – every animal and plant ever eaten is likely unique in terms of their exact protein and metabolite profile depending upon their unique environmental conditions and stressors.
Unfortunately there are a number of experimental design problems with the Mesnage et al. (2016) paper that complicate the interpretation of the results, and as concerning there appear to be confounders that further complicate the analyses.
These include:
- Only a single replicate of each treatment (n=1) at a single location (over two years) is analyzed with no biological replication or randomization of locations to remove site variability.
- The data from the two cultivations in different years were inexplicably merged prior to analysis which made it impossible to determine if results or trends were consistent or reproducible between years
- No inclusion of non-GM reference-varieties (conventional commercial hybrids) representative of those that would be normally grown in the areas where the field trials are performed to put some figures and context to the natural biological variation in the composition of non-GM corn comparators
- No discussion of correction for multiple comparisons (by chance one in every 20 comparisons would be expected to be significant at the p<0.05) of so. If doing multiple comparisons it is necessary to do a multiple-comparison correction
- There appears to be evidence of different levels of fungal (Gibberella moniliformis Maize ear and stalk rot fungus) protein contamination between the three groups. See Supplemental Dataset 5 where Tubulin alpha chain OS=Gibberella moniliformis (strain M3125 / FGSC 7600) appears as the protein that had the biggest fold change between control and GM lines. If there were differing levels of fungal infestation among the groups this would also confound the data.
Others have commented on some of their concerns with this paper including a comprehensive analysis from a number of scientists with expertise in this area. There were also comments from European experts from the science media center. And another discussed the definition and importance of true isogenic lines.
Based on significant differences between proteins and metabolites, including the rather alarmingly named “putrescine and cadaverine” which were markedly increased in the GM NK603 corn (N-acetyl-cadaverine (2.9-fold), N-acetylputrescine (1.8-fold), putrescine (2.7-fold) and cadaverine (28-fold), Mesnage et al. (2016) concluded that NK603 and its isogenic control line are not substantially equivalent, meaning that there were statistical differences between the proteins and metabolites found in the three groups. However what is not clear is whether the levels and variation for characteristics in the genetically modified organism or the control were within the natural range of variation for those characteristics in corn, and the biological significance of the statistical differences in terms of posing a food safety concern. Differences between the GM variety in the presence and absence of Roundup would presumably be similar to the differences that occur every time a crop is treated with an herbicide, be the plant GM or not.
I could not resist looking up these two metabolites putrescine and cadaverine which seem like they should more appropriately be associated with a decaying animal corpse. According to Wikipedia, “Putrescine, or tetramethylenediamine, is a foul-smelling organic chemical compound that is related to cadaverine; both are produced by the breakdown of amino acids in living and dead organisms and both are toxic in large doses. The two compounds are largely responsible for the foul odor of putrefying flesh, but also contribute to the odor of such processes as bad breath and bacterial vaginosis. More specifically, cadaverine is a foul-smelling diamine compound produced by the putrefaction of animal tissue.”
So what are these two horrifying compounds doing in corn samples? Enquiring minds needed to know. So being a good scientist I googled “Cadaverine in corn”, and lo and behold a peer-reviewed study. Check out Table 1. Mean levels of free bioactive amines in fresh, canned and dried sweet corn (Zea mays).
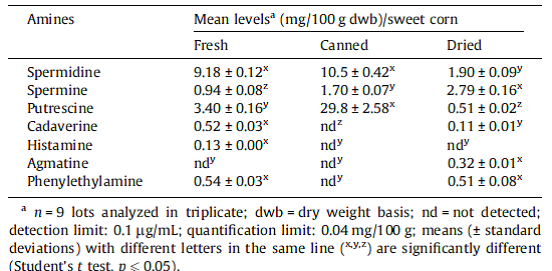
According to this study on “Bioactive amines in fresh, canned and dried sweet corn, embryo and endosperm and germinated corn”, “Different levels of amines in corn products were reported in the literature. Okamoto et al. (1997) found higher concentrations of putrescine and spermidine in fresh corn. Zoumas-Morse et al. (2007) reported lower spermidine and putrescine levels in fresh and canned corn. The differences observed on the profile and levels of amines may be related to several factors such as cultivars, cultivation practices, water stress, harvest time, grain maturity, types of processing and storage time.” In other words, there is a lot of natural biological variation in the different plant varieties we commonly consume with regard to the amount of amines in corn products, and yet we commonly and safely consume fresh, canned and dried sweet corn. If you really want to get nerdy, there are databases of polyamines in food.
As the multi-omics analysis of the NK603 Roundup-tolerant GM maize paper by Mesnage correctly states “the vagueness of the term substantial equivalence generates conflict amount stakeholders to determine which compositional differences are sufficient to declare a GMO as non-substantially equivalent.” In the absence of knowledge of the natural variation in proteins and metabolites in the common foods we eat, the level of different proteins and metabolites that trigger a safe/unsafe determination, and a testable hypothesis at the outset of an experiment, undisciplined “-omics” studies risk becoming statistical fishing trips.
As someone who works in genomics and knows the tens or even hundreds of thousands of statistical comparisons that are part of genomic analyses, there is a real need to understand the statistical methods required for multiple comparisons. If 10,000 comparisons are made at the p<0.05 rate, 500 would be expected to be statistically significant by chance alone. The biological relevance of statistical differences is also not always clear as discussed here. According to the European Food Safety Authority (EFSA) Scientific Committee, good experimental design requires that “the nature and size of biological changes or differences seen in studies that would be considered relevant should be defined before studies are initiated. The size of such changes should be used to design studies with sufficient statistical power to be able to detect effects of such size if they truly occurred.”
In the first line of the discussion Mesnage et al. state “In this report we present the first multi-omics analysis of GM NK603 maize compared to a near isogenic non-GM counterpart”. There are actually two relevant papers on the NK603 line here and here that were published in 2016 but which were inexplicably not even cited in the Mesnage publication. The later paper is entitled “Evaluation of metabolomics profiles of grain from maize hybrids derived from near-isogenic GM positive and negative segregant inbreds demonstrates that observed differences cannot be attributed unequivocally to the GM trait” which compared differences in grain from corn hybrids derived from a series of GM (NK603, herbicide tolerance) inbreds and corresponding negative segregants. The authors concluded “Results demonstrated that the largest effects on metabolomic variation were associated with different growing locations and the female tester. They further demonstrated that differences observed between GM and non-GM comparators, even in stringent tests utilizing near-isogenic positive and negative segregants, can simply reflect minor genomic differences associated with conventional back-crossing practices.”
Moreover, a 2013 meta-analysis by Ricroch examined data from 60 high-throughput ‘-omics’ comparisons between GE and non-GE crop lines. There are several papers on compositional data in GE versus non-GM corn varieties (here, here, here, here, here, here, here, here). The overwhelming conclusion that is common to these papers is that natural variation due to varying genetic backgrounds and environmental conditions explained most of the variability among the samples. And yet this nuance is missing in the 2016 Mesnage paper – the conflation of any factors other than the genetic modification and treatment with Roundup that could influence the results given the poor experimental design is ignored. This tends to be a common feature of this research group – to ignore standard experimental design protocols such as randomization and biological replication, cherry pick cited literature and ignore contradictory or preceeding studies with dissimilar results, rather than discussing their results in the context of what is known based on the entire weight-of-evidence in the scientific literature.
Ricroch in her meta-analysis summarized that “The ‘-omics’ comparisons revealed that the genetic modification has less impact on plant gene expression and composition than that of conventional plant breeding. Moreover, environmental factors (such as field location, sampling time, or agricultural practices) have a greater impact than transgenesis. None of these ‘-omics’ profiling studies has raised new safety concerns about GE varieties”
Interestingly, one study showed that transcriptome alteration was greater in mutagenized plants than in transgenic plants. Of course the random mutations associated with mutation breeding undergo no regulatory evaluation or substantial equivalence assessment prior to commercialization. Variation is the driver of breeding programs, and the reason that varieties like red delicious and golden delicious apples differ from each other in the first place.
Finally Mesnage et al. acknowledge funding from “The Sustainable Food Alliance” for their paper. There is no link as to which groups or interests provide funding for this Alliance. This is not reassuring and runs counter to the absolute transparency of all funding sources that is being demanded of public sector researchers working in this field.
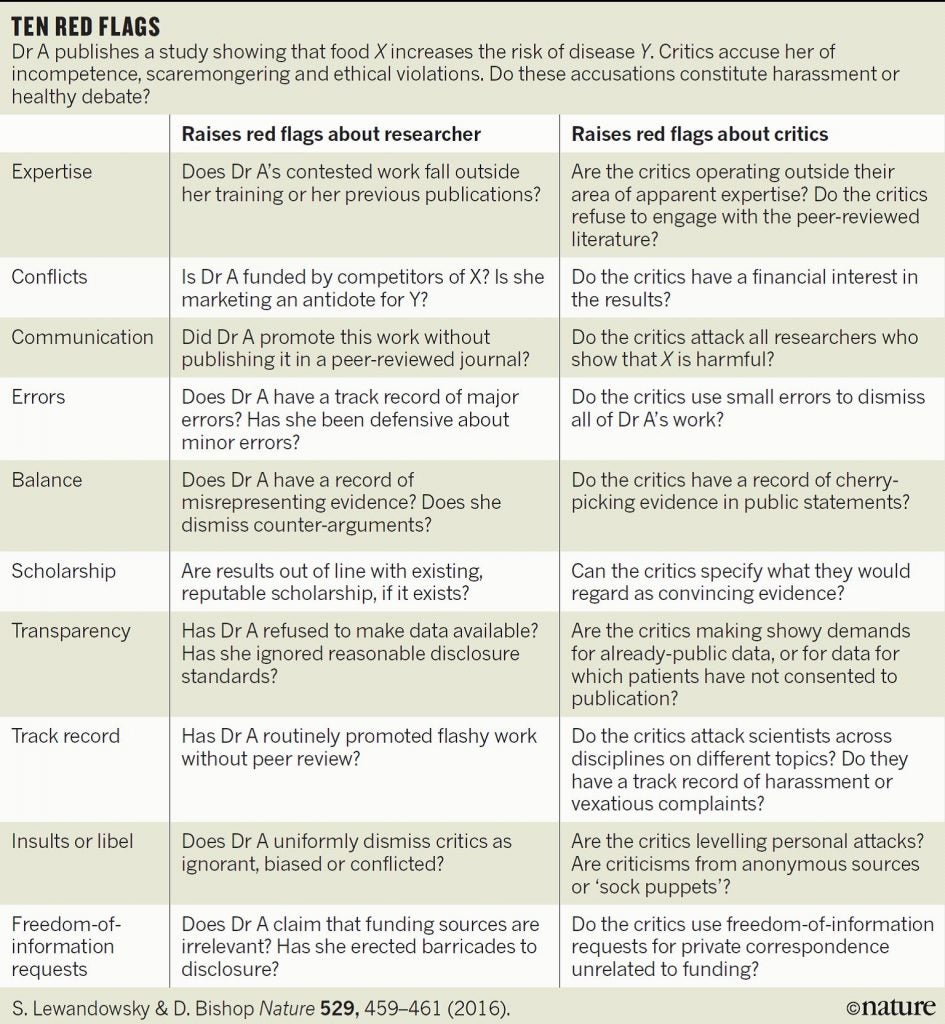
At the end of the day if I have concerns about a paper by a group that has a track record of publishing highly controversial studies, I like to go back to the Nature graphic shown above to see how many red flags are raised. In this case there were a few, most particularly around experimental design and omitting references and discussion of the finding of other “-omics” studies which have consistently shown the high levels of natural variation that is seen in the composition of food due to the differing environments experienced by the plants (and animals) we consume.
I know that this is more of a response than any journalist could ever use, but as with most everything in agriculture, there are no simple sound-bite answers . Having said that I appreciate the press reaching out to seek comment from scientists and hope that is increasingly common in 2017. Although taking the time to respond kinda took the rest of my day. I may have to rethink my New Year’s Resolutions if I plan to get any of my own research done this year, I will worry about that tomorrow when I return to work for the year.